
 Paracetamol toxicity is caused by excessive use or overdose of the analgesic drug paracetamol (called acetaminophen in North America). Mainly causing liver injury, paracetamol toxicity is one of the most common causes of poisoning worldwide. In the United States and the United Kingdom it is the most common cause of acute liver failure.
Paracetamol toxicity is caused by excessive use or overdose of the analgesic drug paracetamol (called acetaminophen in North America). Mainly causing liver injury, paracetamol toxicity is one of the most common causes of poisoning worldwide. In the United States and the United Kingdom it is the most common cause of acute liver failure.Many individuals with paracetamol toxicity may have no symptoms at all in the first 24 hours following overdose. Others may initially have nonspecific complaints such as vague abdominal pain and nausea. With progressive disease, signs of liver failure may develop; these include low blood sugar, low blood pH, easy bleeding, and hepatic encephalopathy. Some will spontaneously resolve, although untreated cases may result in death.
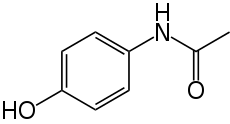 Damage to the liver, or hepatotoxicity, results not from paracetamol itself, but from one of its metabolites, N-acetyl-p-benzoquinoneimine (NAPQI). NAPQI depletes the liver's natural antioxidant glutathione and directly damages cells in the liver, leading to liver failure. Risk factors for toxicity include excessive chronic alcohol intake, fasting or anorexia nervosa, and the use of certain drugs such as isoniazid.
Damage to the liver, or hepatotoxicity, results not from paracetamol itself, but from one of its metabolites, N-acetyl-p-benzoquinoneimine (NAPQI). NAPQI depletes the liver's natural antioxidant glutathione and directly damages cells in the liver, leading to liver failure. Risk factors for toxicity include excessive chronic alcohol intake, fasting or anorexia nervosa, and the use of certain drugs such as isoniazid.Treatment is aimed at removing the paracetamol from the body and replacing glutathione. Activated charcoal can be used to decrease absorption of paracetamol if the patient presents for treatment soon after the overdose; the antidote acetylcysteine acts as a precursor for glutathione, helping the body regenerate enough to prevent damage to the liver. A liver transplant is often required if damage to the liver becomes severe. Patients treated early have a good prognosis, whereas patients that develop major liver abnormalities typically have a poor outcome. Efforts to prevent paracetamol overdose include limiting individual sales of the drug and combining paracetamol with methionine, which is converted into glutathione in the liver.
Toxicity
The toxic dose of paracetamol is highly variable. In general the recommended maximum daily dose for healthy adults is 4 grams. Higher doses lead to increasing risk of toxicity. In adults, single doses above 10 grams or 200 mg/kg of bodyweight, whichever is lower, have a reasonable likelihood of causing toxicity. Toxicity can also occur when multiple smaller doses within 24 hours exceeds these levels. Following a normal dose of 1 gram of paracetamol four times a day for two weeks, patients can expect an increase in alanine transaminase in their liver to typically about three times the normal value. It is unlikely that this dose would lead to liver failure. Studies have shown significant hepatotoxicity is uncommon in patients who have taken greater than normal doses over 3 to 4 days. In adults, a dose of 6 grams a day over the preceding 48 hours could potentially lead to toxicity, while in children acute doses above 200 mg/kg could potentially cause toxicity. Acute paracetamol overdose in children rarely causes illness or death, and it is very uncommon for children to have levels that require treatment, with chronic larger-than-normal doses being the major cause of toxicity in children. Intravenous doses should be smaller than those taken orally, all other things being equal.In rare individuals, paracetamol toxicity can result from normal use. This may be due to individual ("idiosyncratic") differences in the expression and activity of certain enzymes in one of the metabolic pathways that handle paracetamol.
Signs and symptoms
The signs and symptoms of paracetamol toxicity occur in three phases. The first phase begins within hours of overdose, and consists of nausea, vomiting, pallor, and sweating. However, patients often have no specific symptoms or only mild symptoms in the first 24 hours of poisoning. Rarely, after massive overdoses, patients may develop symptoms of metabolic acidosis and coma early in the course of poisoning.The second phase occurs between 24 and 72 hours following overdose and consists of signs of increasing liver damage. In general, damage occurs in hepatocytes as they metabolize the paracetamol. The individual may experience right-upper-quadrant pain. The increasing liver damage also alters biochemical markers of liver function; International normalized ratio (INR) and the hepatic transaminases alanine transaminase and aspartate transaminase rise to abnormal levels. Acute kidney failure may also occur during this phase, typically caused by either hepatorenal syndrome or multiple organ dysfunction syndrome. In some cases, acute kidney failure may be the primary clinical manifestation of toxicity. In these cases, it has been suggested that the toxic metabolite is produced more in the kidneys than in the liver.
The third phase follows at 3 to 5 days, and is marked by complications of massive hepatic necrosis leading to fulminant hepatic failure with complications of coagulation defects, hypoglycemia, kidney failure, hepatic encephalopathy, cerebral edema, sepsis, multiple organ failure, and death. If the third phase is survived, the hepatic necrosis runs its course, and liver and kidney function typically return to normal in a few weeks. The severity of paracetamol toxicity varies depending on the dose and whether appropriate treatment is received.
Pathophysiology
When taken in normal therapeutic doses, paracetamol has been shown to be safe. Following a therapeutic dose, it is mostly converted to nontoxic metabolites via Phase II metabolism by conjugation with sulfate and glucuronide, with a small portion being oxidized via the cytochrome P450 enzyme system. Cytochromes P450 2E1 and 3A4 convert approximately 5% of paracetamol to a highly-reactive intermediary metabolite, N-acetyl-p-benzoquinoneimine (NAPQI). Under normal conditions, NAPQI is detoxified by conjugation with glutathione to form cysteine and mercapturic acid conjugates.
In cases of paracetamol overdose, the sulfate and glucuronide pathways become saturated, and more paracetamol is shunted to the cytochrome P450 system to produce NAPQI. As a result, hepatocellular supplies of glutathione become depleted, as the demand for glutathione is higher than its regeneration. NAPQI therefore remains in its toxic form in the liver and reacts with cellular membrane molecules, resulting in widespread hepatocyte damage and death, leading to acute hepatic necrosis. In animal studies, hepatic glutathione must be depleted to less than 70% of normal levels before hepatotoxicity occurs.

Risk factors
A number of factors can potentially increase the risk of developing paracetamol toxicity. Chronic excessive alcohol consumption can induce CYP2E1, thus increasing the potential toxicity of paracetamol. Whether chronic alcoholism should be considered a risk factor has been debated by some clinical toxicologists.For chronic alcohol users, acute alcohol ingestion at the time of a paracetamol overdose may have a protective effect. For non-chronic alcohol users, acute alcohol consumption had no protective effect.Fasting is a risk factor, possibly because of depletion of hepatic glutathione reserves. The concomitant use of the CYP2E1 inducer isoniazid increases the risk of hepatotoxicity, though whether 2E1 induction is related to the hepatotoxicity in this case is unclear. Concomitant use of other drugs that induce CYP enzymes, such as antiepileptics including carbamazepine, phenytoin, and barbiturates, have also been reported as risk factors.
According to a preliminary study conducted by the University of Washington, mixing large amounts of both paracetamol and caffeine may cause liver damage. Researchers discovered that caffeine can triple the amount of NAPQI. This reaction can be caused by large doses of over-the-counter pain relief that combine caffeine and paracetamol. Dr. Sid Nelson, a professor of medicinal chemistry at the University of Washington, said, "Caffeine can interact with an enzyme that can form a toxic metabolite of paracetamol in such a way that it increases the formation of that toxic metabolite." However, the amount of caffeine that was shown to cause the effect in the study was an order of magnitude higher than typical doses experienced by coffee drinkers.
Diagnosis
The most effective way to diagnose poisoning is by obtaining a blood paracetamol level. A drug nomogram developed in 1975, called the Rumack-Matthew nomogram, estimates the risk of toxicity based on the serum concentration of paracetamol at a given number of hours after ingestion. To determine the risk of potential hepatotoxicity, the paracetamol level is traced along the nomogram. Use of a timed serum paracetamol level plotted on the nomogram appears to be the best marker indicating the potential for liver injury. A paracetamol level drawn in the first four hours after ingestion may underestimate the amount in the system because paracetamol may still be in the process of being absorbed from the gastrointestinal tract. Therefore a serum level taken before 4 hours is not recommended.Clinical or biochemical evidence of liver toxicity may develop in one to four days, although, in severe cases, it may be evident in 12 hours. Right-upper-quadrant tenderness may be present and can aid in diagnosis. Laboratory studies may show evidence of hepatic necrosis with elevated AST, ALT, bilirubin, and prolonged coagulation times, particularly an elevated prothrombin time. After paracetamol overdose, when AST and ALT exceed 1000 IU/L, paracetamol-induced hepatotoxicity can be diagnosed. In some cases, the AST and ALT levels can exceed 10,000 IU/L.

Prevention
Besides preventing an overdose, one way to prevent liver damage may be the use of Paradote. Paradote is a combination tablet containing 100 mg methionine and 500 mg paracetamol. Methionine is included in order to ensure that sufficient levels of glutathione in the liver are maintained in order to minimize the liver damage caused if a paracetamol overdose is taken.Other attempts at minimizing paracetamol's adverse effects have been suggested including adding an emetic agent to tablets, reducing publicity about paracetamol, the inclusion of warnings on packs of paracetamol, and limiting the quantity of the drug sold. Few of these measures have been tried, as they either are not practical or have potential safety issues that make them unsuitable. Limiting the availability of paracetamol tablets has been attempted in some countries. In the UK, sales of over-the-counter paracetamol are restricted to packs of 32 tablets in pharmacies, and 16 tablets in non-pharmacy outlets. Pharmacists may provide up to 100 tablets for those with chronic conditions at the pharmacist's discretion. In Ireland, the limits are 24 and 12 tablets, respectively. It is unclear whether these interventions actually reduce poisoning deaths from paracetamol overdose.
One suggested method of prevention is to make paracetamol a prescription-only medicine, or to remove it entirely from the market. However, overdose is a relatively minor problem; for example, only 0.08% of the UK population present with paracetamol overdose each year. In contrast, paracetamol is a safe and effective medication that is taken without complications by millions of people. In addition, alternative pain relief medications such as aspirin are more toxic in overdose, whereas non-steroidal anti-inflammatory drugs are associated with more adverse effects following normal use.
Paracetamol ester prodrug with L-pyroglutamic acid (PCA), a biosynthetic precursors of glutathione, has been synthesized to reduce paracetamol hepatotoxicity and improve bioavailability. The toxicological studies of different paracetamol esters show that L-5-oxo-pyrrolidine-2-paracetamol carboxylate reduces toxicity after administration of an overdose of paracetamol to mice. The glutathione hepatic values in mice induced by intraperitoneal injection of the ester are superimposable with the GSH levels recorded in no-treated mice control group. The mice group treated with an equivalent dose of paracetamol showed a significative decrease of gluthathione of 35% (p<0.01 vs untreated control group). The oral LD50 was found to be greater than 2000 mg kg-1, whereas the intraperitoneal LD50 was 1900 mg kg-1. These results taken together with the good hydrolysis and bioavailability data show that this ester is a potential candidate as a prodrug of paracetamol.
Treatment
Acetylcysteine
Acetylcysteine, also called N-acetylcysteine or NAC, works to reduce paracetamol toxicity by replenishing body stores of the antioxidant glutathione. Glutathione react with the toxic NAPQI metabolite so that it does not damage cells and can be safely excreted. Cysteamine and methionine have also been used to prevent hepatotoxicity, although studies show that both are associated with more adverse effects than acetylcysteine. Additionally, acetylcysteine has been shown to be a more effective antidote, particularly in patients presenting greater than 8 hours post-ingestion.If the patient presents less than eight hours after paracetamol overdose, then acetylcysteine significantly reduces the risk of serious hepatotoxicity and guarantees survival. If acetylcysteine is started more than 8 hours after ingestion, there is a sharp decline in its effectiveness because the cascade of toxic events in the liver has already begun, and the risk of acute hepatic necrosis and death increases dramatically. Although acetylcysteine is most effective if given early, it still has beneficial effects if given as late as 48 hours after ingestion. In clinical practice, if the patient presents more than eight hours after the paracetamol overdose, then activated charcoal is not useful, and acetylcysteine is started immediately. In earlier presentations, charcoal can be given when the patient arrives and acetylcysteine is initiated while waiting for the paracetamol level results to return from the laboratory.
In United States practice, intravenous (IV) and oral administration are considered to be equally effective if given within 8 hours of ingestion. However, IV is the only recommended route in Australasian and British practice.[4] Oral acetylcysteine is given as a 140 mg/kg loading dose followed by 70 mg/kg every four hours for 17 more doses. Oral acetylcysteine may be poorly tolerated due to its unpleasant taste, odor, and its tendency to cause nausea and vomiting. If repeat doses of charcoal are indicated because of another ingested drug, then subsequent doses of charcoal and acetylcysteine should be staggered.
Intravenous acetylcysteine is given as a continuous infusion over 20 hours for a total dose 300 mg/kg. Recommended administration involves infusion of a 150 mg/kg loading dose over 15 to 60 minutes, followed by a 50 mg/kg infusion over four hours; the last 100 mg/kg are infused over the remaining 16 hours of the protocol. Intravenous acetylcysteine has the advantage of shortening hospital stay, increasing both doctor and patient convenience, and allowing administration of activated charcoal to reduce absorption of both the paracetamol and any co-ingested drugs without concerns about interference with oral acetylcysteine.
The most common adverse effect to acetylcysteine treatment is an anaphylactoid reaction, usually manifested by rash, wheeze, or mild hypotension. Adverse reactions are more common in people treated with IV acetylcysteine, occurring in 4 to 23% of patients. Rarely, severe life-threatening reactions may occur in predisposed individuals, such as patients with asthma. If a anaphylactoid reaction occurs the acetylcysteine is temporarily halted or slowed and antihistamines and other supportive care is administered.

